

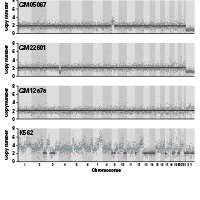
Normal cells can be transformed into malignant tumor cells by acquiring a series of somatic mutation events, such as copy number variations (CNVs). CNVs are extremely common in tumors and are also well-known for driving tumor progression. To identify actionable biomarkers and formulate optimal treatment strategies, we need to understand the driving mutations in individual cells of tumor tissues. Single-cell DNA-seq (scDNA-seq) was developed to answer these scientific questions. However, current scDNA-seq methods for CNV profiling have several major drawbacks. Targeted panels can profile single-cell CNVs in a high-throughput manner, but they lack whole-genome coverage and cannot be used for the detection of arm-level CNVs. Existing methods with whole-genome coverage have not been scaled up to satisfy the need for high-throughput testing. To address these limitations, we have developed the Shasta Whole-Genome Amplification (Shasta WGA) Kit, which has a workflow based on our gold-standard PicoPLEX technology (Figure 1, Panel A). Libraries generated have high rates of mapping and unique reads (Figure 1, Panel B). Shasta WGA enables single-cell CNV profiling with whole-genome coverage for up to 1,500 single cells per experiment. This high-throughput method, which has been optimized on our Shasta Single-Cell System, is paired with key updates to the freely available Cogent NGS tools, enabling you to analyze your scDNA-seq data seamlessly and comprehensively.

Figure 1. Shasta WGA workflow. Panel A. Library preparation begins with (1) a single-cell suspension, which the Shasta instrument dispenses into a 5,184-nanowell chip. After imaging and selection of nanowells containing single cells, (2) the Shasta system dispenses reagents for preamplification and amplification steps, followed by (3) dispensing of 72 i5 and 72 i7 primers for on-chip barcoding PCR. (4) After off-chip purification, the pooled, barcoded libraries are ready for Illumina® sequencing. Finally, (5) sequencing and data analysis are performed. Panel B. QC metrics of adapter Shasta WGA libraries generated from single cells of four different cell lines. For barcoded single cells, after trimming, 99.12% of the barcoded reads were unique reads, and 94.06% of the barcoded reads were uniquely mapped to the hg38 reference genome.


