CRISPR/Cas9 is a revolutionary tool that offers simplicity and flexibility for gene editing experiments. Stem cells are ideal candidates for editing, as they are highly renewable and expandable, and they can differentiate into multiple cell types. Human induced pluripotent stem (hiPS) cells offer the greatest utility due to their practically unlimited expansion and differentiation potential.
The sequential processes of somatic cell reprogramming to create patient-specific hiPS cells, CRISPR/Cas9 gene editing, and single-cell cloning uniquely enable researchers to study how a specific genetic modification can influence function. To create cell models for the discovery of disease etiology, progression, and treatment, isogenic cell lines can be created from healthy or sick individuals, then compared (Figure 1).
Figure 1. Human iPS cells offer a renewable source of clonal diseased or healthy cells.
Cells from a healthy individual can be reprogrammed to create an expandable population of healthy hiPS cells. This healthy hiPS cell population can be edited to insert a known or theorized mutation and expanded clonally to yield complementary cell line(s) that only differ from the "healthy" cells by the introduced mutation. Later, this edited clonal cell line can be differentiated into a cell type relevant to the disease under study (e.g., neurons, as depicted in Figure 1). Alternatively, cells from a sick individual can be reprogrammed and expanded into a "diseased" hiPS cell line, then gene editing can be used to correct the mutation thought to cause the disease. These edited clonal cell lines can be differentiated into the cell type of interest as well. These renewable sources of diseased and healthy cell models, controlled for genetic variability, can be used for a variety of downstream applications to study and treat disease.
Challenges of editing hiPS cells and novel solutions
Despite the power and utility of CRISPR/Cas9 as an editing tool, some challenges must be overcome, particularly when editing hiPS cells. Delivery of Cas9 and the target-specific sgRNA requires an efficient method with low toxicity. Off-target effects should be minimized by the experimental design: choose a good sgRNA with the fewest possible off-target effects, control the amount of Cas9 introduced into the cells, and limit the time that Cas9 is present in the cells. Once edited, single cells with the desired mutation need to be expanded clonally, an inefficient process. Throughout all stages of editing and single-cell-expansion experiments, hiPS cells need to be highly proliferative and pluripotent.
To address the editing difficulties, we have developed a high-quality recombinant Cas9 protein, purified from E. coli, that is ready for use in electroporation experiments. Thus, there is no persistent expression of Cas9, because no coding gene is present. Combined with sgRNAs produced with our Guide-it In Vitro Transcription Kit, we consistently achieve high levels of functional gene knockouts and homology-directed repair (HDR) in hard-to-edit cells, including hematopoietic stem cells (HSCs) and hiPS cells.
To overcome the challenges with editing and single-cell cloning of hiPS cells, we have developed a method that combines the Cellartis iPSC Single-Cell Cloning DEF-CS Culture Media Kit with Guide-it Recombinant Cas9 (10 µg/µl). This method utilizes the DEF-CS culture system, recognized for its suitability for genome engineering (Valton et al. 2014) and single-cell cloning (Feng et al. 2014), to promote reliable growth of hiPS cells in a feeder-free and defined environment.
With this system, hiPS cells are grown as a homogeneous monolayer and are enzymatically passaged as single cells that maintain pluripotency with a stable karyotype for more than 20 passages (Asplund et al. 2016). The same reagents are used for monolayer and single-cell culture, reducing variability. When plated as single cells in wells of a 96-well plate, at least 50% of those seeded cells will proliferate into clonal colonies. In the following experiments, we demonstrate that the Cellartis iPSC Single-Cell Cloning DEF-CS Culture Media Kit combined with Guide-it Recombinant Cas9 (10 µg/µl) is ideal for performing footprint-free gene editing and single-cell cloning of edited hiPS cells.
Results
Maintenance of pluripotency after editing: AcGFP1 knockout test case
An initial proof-of-concept experiment was performed to confirm that editing hiPS cells using electroporation does not influence pluripotency (Figure 2, Panel A). First, cells from Cellartis Human iPS Cell Line 22 (ChiPSC22) were modified to stably express AcGFP1, then these cells were cultured using the DEF-CS culture system in a 48-well plate. Next, we electroporated Guide-it Recombinant Cas9 in a ribonucleoprotein (RNP) complex with sgRNA specific to AcGFP1. The cells were then grown for nine days according to the Cellartis DEF-CS 500 Culture System User Manual. After, cells were analyzed by flow cytometry or immunocytochemistry for AcGFP1 and pluripotency markers. Untreated cells (negative control) were grown in parallel for the same amount of time as treated cells.
To determine pluripotency and AcGFP1 knockout efficiency in electroporated cells, cells were labeled with a fluorescently labeled antibody specific to the pluripotency markers Oct-4 and SSEA-4. Cells were analyzed via flow cytometry and the percentages of cells that were AcGFP1 positive Oct-4 positive, and SSEA-4 positive were quantified (Figure 2, Panel B). The negative control hiPS cells were ~94% AcGFP1 positive, ~96% Oct-4 positive, and over 99% SSEA-4 positive, indicating the cells were unedited and pluripotent. Treatment of hiPS cells by electroporating Cas9/sgRNA RNP complexes targeting AcGFP1 resulted in knockout of AcGFP1 expression in 90% of the cells. Furthermore, pluripotency-determined by Oct-4 and SSEA-4 expression-was maintained in 92% and 99% of analyzed cells, respectively.
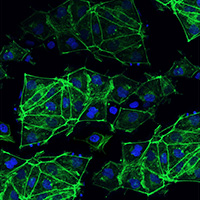
Additional confirmation of knockout and pluripotency was performed using immunocytochemistry (Figure 2, Panel C). Control- and electroporated hiPS cells were assessed for AcGFP1 expression (green), labeled with an antibody specific to Oct-4, visualized with a fluorescent-labeled secondary antibody (red), and nuclear-labeled with DAPI (blue). An isotype control for the secondary antibody was used as a negative control in both samples. In accordance with the flow cytometry data, expression of AcGFP1 in gesicle-treated cells was knocked out in the majority of cells, despite AcGFP1 being expressed in nearly all control cells. Nearly all hiPS cells were Oct-4 positive, supporting the conclusion that editing using electroporation does not alter pluripotency.
Figure 2. Electroporation knockout of AcGFP1 hiPS cells does not alter pluripotency when performed in the DEF-CS culture system. Panel A. Electroporation delivery of Cas9/sgRNA ribonucleoproteins (RNPs) was used to knockout AcGFP1 in the Cellartis Human iPS Cell Line 22 stably expressing AcGFP1. Cells were cultured under non-differentiating conditions using the Cellartis DEF-CS 500 Culture System. After editing, cells were analyzed for AcGFP1 expression and pluripotency via flow cytometry (Panel B) and immunocytochemistry (Panel C). As a negative control, untreated parental cells were grown in parallel for the same amount of time. The quantification of AcGFP1 expression in the edited cells revealed a 90% knockout. Furthermore, pluripotency of the parental and edited cell populations was maintained, with nearly 100% of cells expressing SSEA-4 (assessed via flow cytometry in Panel B) and Oct-4 (assessed via flow cytometry in Panel B and immunohistochemistry in Panel C).
Workflow for generating clonal hiPS cell lines deficient in CD81
After concluding from our proof-of-concept experiment that a successful electroporation-based knockout of a virally integrated gene can be achieved, we chose to target CD81, an endogenous membrane glycoprotein that forms complexes with integrins and plays a critical role in the infection process that leads to hepatitis C (Figure 3).
Figure 3. Workflow for targeted knockout of CD81, an endogenous gene in hiPS cells, using the Cellartis iPSC Single-Cell Cloning DEF-CS Culture Media Kit combined with Guide-it Recombinant Cas9 (10 µg/µl). hiPS cells can be cultured, edited, and clonally expanded using the Cellartis iPSC Single-Cell Cloning DEF-CS Culture Media Kit combined with Guide-it Recombinant Cas9 (10 µg/µl). Before and after using this system, hiPS cells are grown in the DEF-CS culture system, which maintains cells as a karyotypically stable and pluripotent monolayer.
Knockout of CD81 in cells from Cellartis Human iPS Cell Line 18 (ChiPSC18) was performed according to the Cellartis iPSC rCas9 Electroporation and Single-Cell Cloning System User Manual. Electroporation of Cas9 in an RNP complex with sgRNA specifically targeting CD81 was performed on the cells. Flow cytometry analysis of the total population was performed seven days later to determine the CD81 KO efficiency and overall pluripotency levels. Flow cytometry was employed to isolate the sorted population of CD81-negative cells. Single cells from this sorted population were then seeded into each well of a 96-well plate by limiting dilution. Single hiPS cells were expanded into clonal lines using the Cellartis iPSC Single-Cell Cloning DEF-CS Culture Media Kit and characterized for their pluripotency and karyotype.
Successful editing of human iPS cells and maintenance of pluripotency
After electroporation, assessment of CD81 expression by flow cytometry identified that 86.8% of electroporated hiPS cells were CD81 negative (Figure 4, Panel A). We also interrogated the population of sorted, CD81-negative cells for the expression of pluripotency markers (Figure 4, Panel B) and identified that successfully edited hiPS cells were 96.4% Oct-4 positive and 99.9% SSEA-4 positive. Taken together, these data indicate that hiPS cells expanded in the DEF-CS culture system can be successfully edited using electroporation while retaining their pluripotency.
Figure 4. CD81-negative hiPS cells remain pluripotent in the DEF-CS culture system. Panel A. Electroporation of Cas9/sgRNA RNPs was used to knockout CD81 in the Cellartis Human iPS Cell Line 18. Cells were cultured under non-differentiating conditions using the Cellartis DEF-CS 500 Culture System. After editing, cells were analyzed for CD81 expression via flow cytometry. As a negative control, unedited parental cells were grown in parallel for the same amount of time. Quantification of CD81 expression in the electroporated cells revealed an 86.8% knockout efficiency. Panel B. CD81-negative cells were examined for pluripotency markers Oct-4 and SSEA-4. Pluripotency of parental (negative control) and edited (CD81–) cell populations was maintained, with 96.4% of edited cells expressing Oct-4 and ~100% of edited cells expressing SSEA-4.
Following electroporation, CD81-negative hiPS cells sorted via flow cytometry were counted using a hemocytometer. Cells were seeded as single cells using either limited dilution or flow cytometry. For flow cytometry, a single-cell was seeded into each well of a 96-well plate. Limiting dilution was performed by serial dilution until a final theoretical concentration of 50 cells/9,600 µl was achieved. Next, 100 µl of the final cell suspension was added to each well of a 96-well plate (reaching a final theoretical concentration of 0.5 cells/well). This increased the likelihood of obtaining wells containing only single cells and minimized the existence of doublets, which prevent the derivation of a clonal population. Following two weeks of culture, we identified between 31 and 33 emerging colonies from the single-cell seeding of ~50 cells using limited dilution, giving a calculated single-cell survival efficiency of ~62–66% (Table 1). hiPS cells seeded using flow cytometry had a slightly lower survival (52/96, 54%) likely due to the increased stress of passing through the flow cytometer into the plate. However, since seeding with the flow cytometer enabled more single cells to be plated, the overall number of emerging colonies was increased. These data demonstrate that the Cellartis iPSC rCas9 Electroporation and Single-Cell Cloning System enables highly efficient survival of single hiPS cells.
ChiPSC cell line
Isolation method
Emerging colonies at 2 weeks from single cell
Theoretical # of clones plated
Calculated single-cell survival
ChiPSC 22
Limiting dilution
31
50
62%
ChiPSC 18
FACS
52
96
54%
ChiPSC 18
Limiting dilution
33
50
66%
Table I. Highly efficient survival of edited clones grown using the Cellartis iPSC Single-Cell Cloning DEF-CS Culture Media Kit. Typical rates for the generation of clonal colonies from a single-cell cloning experiment range from 1–5%. Using the Cellartis iPSC Single-Cell Cloning DEF-CS Culture Media Kit, we achieved an unparalleled survival rate of 54–66%.
Maintenance of pluripotency in expanded hiPS cell clones
Twelve of the emerging colonies from above were selected and expanded according to the user manual for the Cellartis iPSC rCas9 Electroporation and Single-Cell Cloning System. Once scaled up, each individual clonal line was assessed for pluripotency and knockout of CD81 using flow cytometry (Figure 5). All clones expressed high levels of Oct-4 (97–99% positive), TRA-1-60 (98–99% positive), and SSEA-4 (98–99% positive). Moreover, all lines were found to be CD81 deficient. These data show that pluripotency markers in the expanded clones are maintained at levels comparable to those in the parental line, ChiPSC18. Thus, using the Cellartis iPSC Single-Cell Cloning DEF-CS Culture Media Kit combined with Guide-it Recombinant Cas9 (10 µg/µl), we successfully knocked out endogenous CD81 from a starting line and generated 12 new, edited lines that were still pluripotent.
Figure 5. Pluripotency was maintained in edited hiPS cell clones that were seeded as single cells. Individual, edited (CD81 knockout) hiPS cells were expanded into clonal lines and analyzed for expression of CD81 and three pluripotency markers via flow cytometry using antibodies against CD81, Oct-4, TRA-1-60, and SSEA-4. The parental hiPS cell line, ChiPSC18, was used as a control. As expected, all edited clones exhibited the loss of CD81 expression. Pluripotency was maintained in all edited clonal lines, as evidenced by the persistent expression of the three pluripotency markers.
Occurrence of a diverse set of indels in hiPS cell clones from the CD81 knockout experiment
We examined the specific base-pair (bp) insertions and deletions (indels) created during the CRISPR/Cas9 editing process in the clones using the Guide-it Indel Identification Kit. Because Cas9-induced double-strand breaks are mainly repaired via the error-prone, non-homologous-end-joining (NHEJ) DNA repair pathway, every cell that is edited should have a unique set of indels at the targeted gene-in this experiment, CD81. Accordingly, we saw a wide range of indels in the different clonal cell lines (Figure 6 and Table II). In some cases, CD81 knockout was accomplished via relatively small indels. For example, clone #1 had only a 1-bp insertion on both alleles at the editing site, and clone #10 had only a 1-bp deletion on both alleles at the editing site. Conversely, some indels were much larger. For example, clone #9 had an 53-bp insertion on one allele and a 51-bp insertion on the other allele. Taken together, these data demonstrate the diversity of indels created by CRISPR/Cas9 editing and highlight the utility in creating and screening multiple clones to account for this variability.
Figure 6. Representation of indels created at the CD81 target site in different CRISPR/Cas9-edited hiPS cell clones. The genomic CD81 region of each clone was PCR amplified and sequenced using the Guide-it Indel Identification Kit to obtain detailed sequence information. The results show that each clone has a unique set of biallelic indels that knockout CD81. See Table II for a tabular representation of these data.
Diversity of indels in CRISPR/Cas9 gesicle-treated hiPS cell clones
Clone #
1
2
3
4
5
6
7
8
9
10
11
12
Indels (bp)
Allele 1
+1
+3/ –26
–1
–1
–17
–21
–4
–8
+53
–1
–1
+1/–3
Allele 2
+1
–7
–7
+1
+1
–1
–18
–1
+51
–1
–11
–2
Table II. Tabular representation of the sizes and locations of indels created at the target site in CD81 for different CRISPR/Cas9-edited hiPS cell clones. Examples from this dataset are also shown in Figure 6. The results show that each clone has a unique set of indels that knockout CD81. For each allele, "+" values indicate the number of inserted nucleotides, while "–" values indicate the number of deleted nucleotides. If an insertion and deletion happened simultaneously, it is marked with a slash. If the insertion sequence corresponds to another part of the genome, that chromosome is shown between brackets.
Normal karyotypes observed in edited hiPS cells
CRISPR/Cas9-mediated editing and single-cell cloning can be harsh on hiPS cells. Traditionally, these processes can force selective pressures that favor unintended mutations to the karyotype, conferring competitive advantages to in vitro growth. These karyotypic abnormalities render the cells unsuitable for study. Consequently, it is essential that the karyotype remains unaltered for multiple passages after editing.
To confirm karyotype stability, we examined four of the clonal lines, each of which had been expanded from one single cell to a confluent line in a 10-cm dish over a period of approximately one month (Figure 7). All lines were found to have normal, stable karyotypes. Thus, these data show that the Cellartis iPSC Single-Cell Cloning DEF-CS Culture Media Kit combined with Guide-it Recombinant Cas9 (10 µg/µl) effectively edits and expands single-cell hiPS cell clones without introducing karyotypic abnormalities.
Figure 7. hiPS cell clones that have been edited via electroporation maintain a stable karyotype after editing and clonal expansion. The karyotypes of four edited clonal cell lines were analyzed. All clonal lines showed the expected 46, XY karyotype of the original ChiPSC18 cell line. Clonal cell line #6 is shown as an example.
Conclusions
Combining the Cellartis iPSC Single-Cell Cloning DEF-CS Culture Media Kit with Guide-It Recombinant Cas9 (10 μg/μl) provides an efficient and effective method to generate clonal lines of edited hiPS cells. The system can enable high editing efficiency, with no discernible effect on hiPS cell health or pluripotency. Single-cell seeding of edited hiPS cells in this system results in high survival of pluripotent, edited clones, yielding a diverse set of edited clonal lines. Critically, the expanded lines maintain a normal karyotype, rendering the cells suitable for further investigation and use in screening and disease modeling.
Methods
Cell culture
Cellartis human iPS cells from the ChiPSC22 line (Figure 2) and the ChiPSC18 line (Figures 3–7 and Table I) were grown in the Cellartis DEF-CS 500 Culture System (Cat. # Y30010) before editing. After single-cell cloning and expansion into 48-well plates with the Cellartis iPSC Single-Cell Cloning DEF-CS Culture Media Kit, clonal colonies were returned to the Cellartis DEF-CS 500 Culture System for further expansion prior to indel analysis (Figure 6 and Table II) and karyotyping (Figure 7). Please refer to the Cellartis DEF-CS 500 Culture System User Manual and the Cellartis iPSC rCas9 Electroporation and Single-Cell Cloning System User Manual for specific culture conditions and protocols.
Labeling of Oct-4 was performed by cell fixation and permeabilization followed by an incubation with anti-Oct-4-PE antibody (BD Pharmingen, Cat. #560186; 20 µl for 1 x 106 cells) in PBS for 30 minutes. Cell labeling of extracellular proteins or markers was performed following standard labeling procedures. In Figure 4, Panel A, cells were incubated for 30 minutes with anti-SSEA-4-PE (BD Pharmingen, Cat. #560128; 20 µl for 1 x 106 cells), anti-TRA-1-60-FITC (BD Pharmingen, Cat. #560380; 20 µl for 1 x 106 cells), or anti-CD81-FITC (BD Pharmingen, Cat. #551108; 20 µl for 1 x 106 cells) antibodies. In Figure 4, Panel B, cells were incubated simultaneously with anti-SSEA-4 and anti-TRA-1-60 antibodies, since they were labeled with different fluorophores (PE and FITC, respectively).
Immunocytochemistry
ChiPSC22 cells were grown in chamber slides until fixation with 4% paraformaldehyde. Cell permeabilization was achieved with 0.5% Triton X-100 for 5 minutes. After washing with PBS and blocking for 30 minutes (IHC/ICC Blocking Buffer - Low Protein; eBioscience, Cat. #00-4953), cells were incubated with anti-Oct-4 antibody (diluted 1:150; eBioscience , Cat. #41-5841) or an IgG2a K isotype control (diluted 1:150; eBioscience , Cat. #41-4321) for one hour. After the incubation period, cells were washed and mounted with anti-fading reagent containing DAPI (VECTASHIELD Antifade Mounting Medium with DAPI; Vector Laboratories, Cat. #H-1200).
Analysis of the karyotypes of ChiPSC18 cell lines was performed by Cell Guidance Systems.
References
Asplund, A. et al. One Standardized Differentiation Procedure Robustly Generates Homogenous Hepatocyte Cultures Displaying Metabolic Diversity from a Large Panel of Human Pluripotent Stem Cells. Stem Cell Rev. Reports12, 90–104 (2016).
Feng, Q. et al. Scalable Generation of Universal Platelets from Human Induced Pluripotent Stem Cells. Stem Cell Reports3, 817–831 (2014).
Valton, J. et al. Efficient strategies for TALEN-mediated genome editing in mammalian cell lines. Methods69, 151–170 (2014).