


4 factors to consider for immune repertoire profiling

Why do immune repertoire sequencing?
Adaptive immunity relies on the ability of the body to recognize and respond to a nearly endless number of molecules, a complex feat achieved through T-cell and B-cell receptors (TCRs and BCRs, respectively). Receptor-antigen interactions are specific, meaning a tremendous amount of diversity in TCRs and BCRs is required to recognize the wide assortment of antigens one might encounter. To this end, the adaptive immune system has evolved a system for somatic diversification, commonly referred to as V(D)J recombination. Ultimately, this process yields cell populations with sufficient TCR and BCR diversity to recognize any peptide imaginable. However, it is this very diversity of receptor sequences that can make it difficult to analyze them, and a myriad of research applications—from examining the mechanisms behind autoimmune diseases and cancers to developing models for immunotherapy—rely on being successful in this first step.
How are immune repertoires analyzed?
Traditionally, immune repertoire profiling has relied on cloning and Sanger sequencing or functional methods for identifying antigen-specificity (e.g., tetramer assays/stains). Although these methods have yielded valuable insights, the continued development of next-generation sequencing (NGS) has dramatically expanded the scale of repertoire profiling research. Along with this added technology in the researcher's toolkit comes several key decision points on how to proceed with your studies.
What factors should I consider when choosing an immune repertoire sequencing approach?
DNA vs. RNA template
Using genomic DNA as a template can make it easier to determine the relative abundance of clonotypes, as each cell only has a single template. However, this comes at a cost. Because each cell has multiple copies of the TCR or BCR transcript, RNA-seq approaches are more sensitive. Thus, using RNA allows for a more comprehensive identification of unique receptor variants, including those that are present in a very small proportion of cells.
Additionally, sequencing mRNA reveals the sequence of expressed receptors that have undergone splicing and post-transcriptional processing, and thus are more likely to yield functional proteins. DNA-based approaches, by contrast, do not identify receptor sequences in their translated forms, and will unavoidably yield many functionally irrelevant sequences. Additionally, template choice can determine your options for NGS library prep (see below).
CDR3 only vs. full-length sequence
Most immune profiling experiments have focused on the CDR3 region of TCR and BCR sequences. The CDR3 region contains the core site of interaction between antigen and receptor and is, therefore, the most variable, so this is not surprising. However, full-length sequencing will include additional regions of the receptor, including CDR1 and CDR2, which play roles in the affinity of antigen-receptor binding and/or downstream signaling. Full-length sequencing also makes it easier to directly clone and express those receptors identified in immune repertoire sequencing studies. This is particularly important for antibody therapy or T-cell therapy applications.
Bulk vs. single-cell analysis
The choice to use bulk or single-cell analysis depends on the goal of your experiment. Generally, analysis of immune repertoire diversity tends to rely on bulk approaches, while studies focused on identifying TCR or BCR sequences specific to a particular antigen use single-cell analysis. Bulk analyses can sample many more sequences in a single experiment but do so at the cost of pairing information. Functional TCRs are made up of paired alpha and beta chains, while BCRs have paired heavy and light chains. Thus, this pairing information is required for the identification of the actual TCR or BCR molecule in question. Since individual T cells or B cells express a single receptor sequence, single-cell analysis makes it possible to capture both the sequence and the pairing information. However, analyzing single cells generally decreases the number of samples that can be analyzed. Researchers often start with bulk analysis and move to single cells after selecting for binding affinity or other phenotypes of interest.
NGS library prep: multiplex PCR vs. 5′ RACE
The most common method of preparing sequencing libraries for immune profiling is multiplex PCR. Multiplex PCR is appropriate for both DNA and RNA templates and generally consists of two rounds of PCR (after cDNA synthesis, if RNA is used as a template). The first round of PCR amplifies the receptor locus and adds known sequences that act as priming sites for the second round, in which sequencing adaptors and indexes are incorporated. However, the priming sites at the 3′ and especially the 5′ end of the first PCR overlap sites with significant sequence diversity, which means many degenerate primers are needed to amplify different TCR/BCR sequences (see the right panel in the figure below). Of course, this can also introduce significant PCR bias where receptor sequences that are more similar to the primers are amplified more efficiently.
Another approach is 5′ RACE (Rapid Amplification of cDNA Ends) combined with semi-nested PCR (see the left panel in the figure below). This method is only appropriate for RNA inputs, but does avoid the issue of bias introduced by multiplex PCR. During cDNA synthesis, the reverse transcriptase (RT) adds nontemplated nucleotides to the 5′ end of the first-strand cDNA. A template-switching oligo complementary to these nontemplated nucleotides then hybridizes to the first-strand cDNA. This enables the RT to switch templates and incorporate an adapter sequence on the 5′ end. The two subsequent rounds of PCR use this adapter sequence instead of relying on degenerate primers within the TCR or BCR cDNA. This greatly reduces PCR bias and ensures the immune repertoire profile reflects the original sample, not the primer design. In addition, the second PCR is semi-nested, which ensures a high on-target rate, thus reducing sequencing costs.

Comparison of two methods for sequencing library preparation for immune repertoire sequencing. 5′ RACE, shown here as part of the overall workflow for our SMARTer TCR and BCR profiling kits, enables the capture of full-length receptor sequences from mRNA without relying on multiple degenerate primers that overlap regions of variability. Multiplex PCR can use DNA or RNA as template, but often requires sample-specific optimization to avoid PCR bias arising from the use of the aforementioned degenerate primers. In the example schematic for a resulting sequencing-ready library, P5 and P7 refer to Illumina flow cell binding sites added by the primers during library preparation. Primers throughout the diagram are shown as left- and right-pointing arrows along the templates.
We have developed several TCR and BCR profiling kits for both human and mouse samples, utilizing the 5′ RACE method described above. To learn more and view data, explore our immune profiling page. If you have any questions about choosing the right kit for your research, please contact our technical support team.
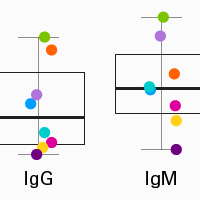
Profiling human B-cell receptors with a 5' RACE approach and SMART technology
The SMARTer Human BCR IgG IgM H/K/L Profiling Kit captures entire V(D)J variable regions of BCR transcripts, allowing you to reach a complete and accurate view of human BCR repertoires. Incorporation of UMIs further improves accuracy and reliability by enabling the removal of reads derived from PCR errors or duplication and/or sequencing errors.
View the data
A SMARTer approach to T-cell receptor profiling
Detect low-abundance TCR variants from human peripheral blood RNA or purified human T cells.
High-throughput TCR profiling with 5'-end differential analysis of single cells
View data on high-throughput whole transcriptome and TCR profiling data from the same cell using the ICELL8 systems.
High-throughput single-cell T-cell receptor profiling with SMART technology
View single-cell TCR profiling data generated with SMART technology and the ICELL8 Single-Cell System.
Takara Bio USA, Inc.
United States/Canada: +1.800.662.2566 • Asia Pacific: +1.650.919.7300 • Europe: +33.(0)1.3904.6880 • Japan: +81.(0)77.565.6999
FOR RESEARCH USE ONLY. NOT FOR USE IN DIAGNOSTIC PROCEDURES. © 2025 Takara Bio Inc. All Rights Reserved. All trademarks are the property of Takara Bio Inc. or its affiliate(s) in the U.S. and/or other countries or their respective owners. Certain trademarks may not be registered in all jurisdictions. Additional product, intellectual property, and restricted use information is available at takarabio.com.
