


Unraveling the world of microorganisms

Microbes are everywhere but, until recently, we were only able to study them in isolation in cultured samples. Metagenomics has opened a whole new era in microbial research; now, we can examine them as a whole in their natural environment.
Metagenomic NGS (mNGS) ups the ante by allowing targeted or untargeted characterization, without bias, of species in mixed populations. Untargeted mNGS uses shotgun sequencing of pure microbial culture samples from which random samples of DNA or RNA are assayed. In contrast, targeted approaches are restricted to the detection of specific species or regions utilizing 16S rRNA amplification, singleplex or multiplex PCR, primer extension, or bait-probe enrichment methods (Wei, Miller, and Chiu 2019).
Shotgun mNGS of patient samples suffers from a 'needle-in-the-haystack' dilemma given the very low pathogen-to-host genome ratio. As >99% of reads using this method are derived from the human host, its pathogen-detection sensitivity is significantly limited. Targeted sequencing or host-depletion methods overcome this hurdle by decreasing the relative proportion of background sequences (host gDNA, rRNA, or mitochondrial RNA) compared to the nonhuman microbial reads. These methods, therefore, offer the potential to improve the analytic sensitivity of pathogen detection while retaining the advantage of unbiased mNGS, since they are agnostic of the pathogen one is seeking.
Untangling the knots of metagenomics methods
In an intriguing study, researchers from the University of Bern, Switzerland, established a straightforward shotgun mNGS workflow to minimize the overwhelming host nucleic acid (NA) background, allowing the simultaneous screening for bacteria and viruses in cerebrospinal fluid (CSF) samples. To attempt to reduce the host NA proportion and enrich viral and bacterial NA within a patient sample, they developed a simple NA depletion method.
The host NA depletion process consisted of selective host cell lysis, enzymatic degradation of the liberated genomic DNA, and a final depletion with paramagnetic beads. The products were subjected to reverse transcription, whole-genome amplification using the PicoPLEX WGA Kit, and then NGS. First, they verified the effectiveness of their workflow on surrogate CSF samples that were spiked with three 1:100 dilutions of Influenza A H3N2 virus. Next, they tested the workflow on surrogate CSF samples spiked with both Influenza A virus and bacteria in order to test the simultaneous detection of RNA and DNA (Figure 1). The workflow was also tested on thirteen CSF samples from meningo-/encephalitis patients (two bacterial, eleven viral etiologies), a serum of an Andes virus infection, and a nose swab of a common cold patient. Host depletion of these patient samples did not result in more reads, indicating the presence of damaged pathogens likely due to the host immune response, but previously diagnosed pathogens were detected in the depleted and the native samples.
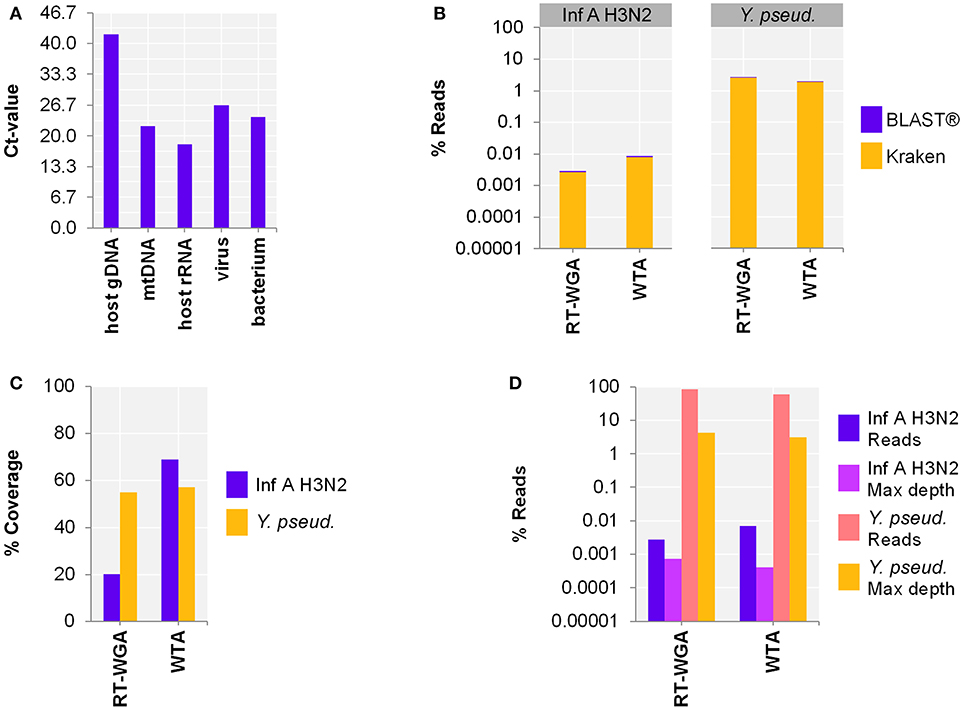
Figure 1. Comparison of amplification approaches after depletion of host nucleic acids (NA) using author's NA-depletion method. Data show a comparison of two detection approaches, whole-transcriptome amplification (WTA) or reverse transcription and whole-genome amplification (RT-WGA), of a host-NA-depleted surrogate CSF sample spiked with RNA virus Influenza A H3N2 (Inf A) and bacteria Y. pseudotuberculosis (Y. pseud). The RT-WGA protocol combined a common reverse transcription kit and the PicoPLEX WGA Kit. Panel A. qPCR results of host-NA-depleted sample. Primer-probe targets are listed on x-axis. The y-axis shows the cycle threshold value (Ct-value), representing an approximate 10-fold reduction in copy number of host NA. Panel B. The metagenomics analysis found that for Inf A, there was a lower percentage of reads with the RT-WGA approach than the WTA approach. The reverse was true for Y. pseud., but to a lesser degree. Panel C. The reference coverage of both Inf A and Y. pseud. was lower after RT-WGA as compared to WTA, while RT-WGA produced a higher maximum depth of coverage (Panel D; pink and yellow bars). Figure adapted from Oechslin et al. 2018 under Creative Commons License (CC BY 4.0).
Pulling the right strings: future directions
Because existing host depletion methods are time-consuming, expensive, and/or highly specialized, the authors of this study looked to develop their own method suitable for liquid clinical samples. The proposed host NA depletion method is straightforward and requires only 10 minutes of hands-on time and 50 minutes of incubation time, and it successfully increases viral and bacterial reads in surrogate CSF samples. When combined with a sensitive whole-genome amplification method, such as the PicoPLEX WGA Kit, it can be a powerful tool to detect bacterial DNA in host samples. While the effectiveness of the technique was encumbered by several factors—including low concentrations of pathogens, numerous contaminations and curation, and the extent of reference databases for bioinformatics analysis—the workflow is a vast improvement over previous methods and, with some tweaks, will pave a way forward for clinical sample analysis.
References
Oechslin, C. P., et al. Limited Correlation of Shotgun Metagenomics Following Host Depletion and Routine Diagnostics for Viruses and Bacteria in Low Concentrated Surrogate and Clinical Samples. Front. Cell. Infect. Microbiol. 8 (2018).
Gu, W., Miller, S., and Chiu, C. Y. Clinical Metagenomic Next-Generation Sequencing for Pathogen Detection. Annu. Rev. Pathol. 14, 319–338 (2019).
Takara Bio USA, Inc.
United States/Canada: +1.800.662.2566 • Asia Pacific: +1.650.919.7300 • Europe: +33.(0)1.3904.6880 • Japan: +81.(0)77.565.6999
FOR RESEARCH USE ONLY. NOT FOR USE IN DIAGNOSTIC PROCEDURES. © 2025 Takara Bio Inc. All Rights Reserved. All trademarks are the property of Takara Bio Inc. or its affiliate(s) in the U.S. and/or other countries or their respective owners. Certain trademarks may not be registered in all jurisdictions. Additional product, intellectual property, and restricted use information is available at takarabio.com.
