CRISPR/Cas9-based gene editing is an effective technique for obtaining knockout mutations with high efficiency, but applying this technology to engineer site-specific insertion of genes or sequences longer than 200 bp via homology-directed repair (HDR) is often very difficult.
A primary challenge in performing CRISPR-based knockins involves achieving efficient delivery of the DNA template which is used for HDR following Cas9-sgRNA-mediated cleavage at the genomic target site. Electroporation of ribonucleoprotein complexes (RNPs) is the preferred method for delivery of Cas9 and sgRNA in vitro due to the easy preparation of the reagents and the transience of their presence in target cells, which limits the possibility of off-target effects. Two popular approaches that leverage this method to engineer knockins involve co-electroporation of the DNA donor template along with the Cas9-sgRNA RNPs, or adeno-associated virus (AAV)-mediated transduction of the donor as ssDNA following RNP electroporation. While the use of AAV often results in high editing efficiencies and on-target specificity, it requires cloning into the appropriate vectors and producing viral particles prior to genome editing.
For approaches involving co-electroporation of the HDR template together with the Cas9-sgRNA RNP, the donor can consist of either double-stranded DNA (dsDNA, linear or plasmid) or single-stranded DNA (ssDNA). However, the application of ssDNA is associated with two important advantages: lower rates of random or off-target integration (Chen et al. 2011, Figure 3; Roth et al. 2018, Extended Figure 4) and lower toxicity (Roth et al. 2018, Extended Figure 1), especially when working with hiPSCs or primary cells.
The Guide-it Long ssDNA Production System v2 provides a simple and fast method for producing long ssDNA (up to 5,000 nt) for use as a donor template in gene editing applications. Below, we discuss the production workflow and show data demonstrating the use of ssDNA to engineer precise, site-specific insertions via HDR.
Preparing long ssDNA with the Guide-it Long ssDNA Production System v2
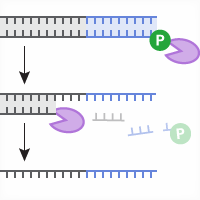
The Guide-it Long ssDNA Production System v2 employs a simple and efficient in vitro protocol for the on-demand generation of long ssDNA strands. The method involves PCR amplification of the donor template using primer pairs that include one phosphorylated oligo. Depending on whether the forward or reverse primer is phosphorylated, the resulting dsDNA PCR product is phosphorylated on either the sense or antisense strand, respectively. In the next step, the phosphorylated strand of the dsDNA is selectively digested by the nucleases Strandase A and B, generating ssDNA. This workflow is summarized by the following schematic:
Figure 1. Preparation of long ssDNA donors using the Guide-it Long ssDNA Production System v2. First, a dsDNA template (insert sequence flanked by 5' and 3' homology arms) is prepared using cloning, fusion PCR, or other related methods. The template should contain arms homologous to the target gene flanking the sequence to be inserted. Next, either of two different dsDNA PCR products is generated from the template using appropriate phosphorylated (P) primers. These PCR products serve as substrates for the strandase reaction: Strandase Mix A (purple enzyme) selectively digests the phosphorylated strand. Next, Strandase Mix B (blue enzyme) is added to finish the digestion and create ssDNA. Finally, the reaction is cleaned up to obtain ssDNA for use as an HDR template in gene knockin experiments.
Results
Production of ssDNA templates
The Guide-it Long ssDNA Production System v2 features improvements that offer two advantages over the original (v1) ssDNA kits (Cat. # 632644, 632645):
A streamlined workflow involving the use of a common buffer for both strandase enzymes
Improved performance over the v1 kit, enabling more efficient processing of challenging templates (Figure 2, Panel A) and higher yields of ssDNA (especially for templates ranging from 500–1,500 nt)
Taking the CCR5 gene as a template for amplifying PCR products of varying lengths, we demonstrated that the kit enables the generation of ssDNA ranging from 500–5,000 nt (Figure 2, Panel B). Production of ssDNA can be confirmed on an agarose gel, with the ssDNA typically running at a lower molecular weight and yielding a band with lower intensity than the corresponding dsDNA PCR substrate.
Figure 2. Improved processing of challenging templates and production of ssDNA up to 5,000 nt with the Guide-it Long ssDNA Production System v2. Panel A. Gel image showing the dsDNA starting material (Lane 1) and sense (SS) or antisense (AS) ssDNA products (Lanes 2 and 3, respectively) for several HDR templates generated using two respective versions of the Guide-it Long ssDNA Production System (v1 or v2). The templates consisted of different inserts flanked by 5′ and 3′ homology arms targeting various genomic loci of interest and had been identified as challenging substrates for ssDNA production using the v1 kit. The dsDNA and ssDNA were analyzed via agarose gel electrophoresis using ethidium bromide as a staining agent. ssDNA products run at a smaller molecular weight than corresponding dsDNA substrates. In all cases, the v2 kit generated a cleaner band of ssDNA than the previous version of the kit, suggesting more complete and uniform digestion. Panel B. The Guide-it Long ssDNA Production System v2 enables successful production of ssDNA ranging from 500 nt to 5,000 nt in length. The gel images show dsDNA substrates of varying length (Lane 1) and corresponding sense (SS) and antisense (AS) ssDNA products (Lane 2) following enzymatic digestion and cleanup with the kit. Each ssDNA HDR template was designed to target the CCR5 gene.
Advantages of ssDNA vs. dsDNA
Reduced random integration
To compare rates of random integration for various types of donor DNA, three different types of templates (plasmid, dsDNA, and ssDNA) designed to fuse AcGFP1 to GADPH were transfected into HEK293 cells alone or in tandem with plasmids encoding Cas9 and an sgRNA targeting GADPH (Figure 3, Panel A). Three days posttransfection, frequencies of GFP-positive cells in the resulting cell populations were determined by flow cytometry (Figure 3, Panel B). The FACS results demonstrated that dsDNA yielded the highest frequency of GFP-positive cells when combined with Cas9 and sgRNA, but that a significant proportion of GFP-positive cells were also produced in the absence of Cas9/sgRNA complexes, suggesting that dsDNA donors can integrate with high efficiency at sites other than a given target site. In contrast, such random, Cas9-independent integration was not observed for ssDNA templates, which efficiently yielded GFP-positive cells only when paired with Cas9 and sgRNA.
Figure 3. Efficient editing and with negligible off-target insertion using ssDNA donor templates. Panel A. Tagging of GAPDH with a fluorescent protein (AcGFP1). An HDR template was designed to fuse the AcGFP1 coding sequence in-frame at the C-terminus of the GAPDH gene. Panel B. HEK293 cells were transfected with plasmid, dsDNA, or ssDNA forms of the AcGFP1 HDR template, with or without cotransfection of plasmids encoding for Cas9 and an sgRNA targeting the GAPDH locus. Cells were grown for three days and then analyzed by flow cytometry. While the plasmid template resulted in very little integration as demonstrated by the low percentages of fluorescent cells, the dsDNA yielded significant proportions of AcGFP1-postive cells, even in the absence of Cas9-sgRNA expression, suggesting a significant level of nonspecific integration. Both sense (S) and antisense (A) ssDNA repair templates were associated with efficient insertion of the AcGFP1 sequence in the presence but not in the absence of Cas9 and sgRNA, suggesting that insertion of the AcGFP1 sequence was highly specific.
Lower cytotoxicity
The effects of dsDNA and ssDNA donor templates on cell viability were also compared following coelectroporation with RNPs in hiPSCs and T cells, respectively (Figure 4, Panels A and B). Visualization of resulting hiPSC populations by microscopy five days postelectroporation revealed that cell counts were dramatically lower following electroporation with dsDNA, as compared to cell populations electroporated with ssDNA (Figure 5, Panel A). Similar results were obtained following electroporation of CD3+ T cells (Figure 5, Panel B). In this case, cell viability was analyzed by flow cytometry 24 hours postelectroporation.
Figure 4. Comparison of cellular toxicity induced by dsDNA and ssDNA upon coelectroporation with Cas9-sgRNA RNPs in different cell types. Panel A. Toxicity of dsDNA in hiPSCs. ChIPSC18 cells were coelectroporated with RNPs and sense or antisense ssDNA or dsDNA donor templates designed to insert AcGFP1 at the locus for the tubulin gene. The donor templates were created with 350-bp homology arms to the insertion site and were 1.5 kb in length. As seen in the images, the dsDNA donor template caused significant toxicity as compared to the ssDNA templates, resulting in lower cell counts three days after electroporation. Panel B. Toxicity of dsDNA in CD3+ T cells. Cells were electroporated with Cas9/sgRNA RNPs with or without HDR templates (dsDNA or ssDNA) designed to insert AcGFP1 at the SEC61B locus. Twenty-four hours after electroporation, cell viability was measured by flow cytometry. Electroporation of dsDNA resulted in higher levels of cell toxicity as compared to ssDNA.
Efficient insertion of an expression cassette into a safe harbor locus in hiPS cells
In a separate experiment, an HDR template was designed for the insertion of an expression cassette encoding AcGFP1 under the constitutive EF1α promoter at the AAVS1 safe harbor site (Figure 5, Panel A) in hiPSCs. The ssDNA produced from the template included 600-nt homology arms with a total length of 3,400 nt, and it was coelectroporated into hiPSCs along with RNPs targeting the AAVS1 locus. Following gene editing, FACS analysis of the resulting cell population showed that 3.64% of the cells were positive for AcGFP1 expression (Figure 6, Panel B), demonstrating efficient knockin of the sizable EF1α-AcGFP1 expression cassette.
Figure 5. Insertion of a fluorescent protein expression cassette into the AAVS1 site in hiPSCs using long ssDNA oligo as donor template. Panel A. Schematic of donor template and Cas9-sgRNA target site for insertion of AcGFP1 expression cassette at the AAVS1 locus. Panel B. FACS analysis of AcGFP1 expression in control and edited populations following electroporation.
Knockin of murine T-cell receptor α- and β-chains at the TRAC locus in primary CD3+ T cells
CAR‐T cells (chimeric antigen receptor-engineered T cells) and TCR‐T cells (T cell receptor-engineered T cells) have emerged as promising therapeutics for hematological and solid malignancies. The application of CRISPR/Cas9 in this field holds great potential, as it enables the insertion of CAR and TCR constructs into specific genomic target sites, and knockout or replacement of endogenous factors (Eyquem et al. 2017).
To demonstrate the applicability of ssDNA for immune cell therapy research, a 2.8-kb HDR template was designed for knockin of murine T-cell receptor α- and β-chains at the TRAC locus in primary human CD3+ T cells using ssDNA as a donor (Schober et al. 2019). Specifically, RNPs with sgRNAs targeting TRAC and TRBC loci, respectively, were electroporated together with the ssDNA HDR template encoding the murine T-cell receptor α- and β-chains. Knockout of both endogenous TCR genes (TRAC and TRBC) was required in order to facilitate sufficient surface expression and ensure correct pairing of the transgenic mouse receptor chains. Ten days after electroporation, flow cytometry was used to detect the successful HDR and expression of the murine TCRs (Figure 6, Panel B).
Figure 6. Knockin of murine T-cell receptor α- and β-chains at the TRAC locus in human CD3+ T cells. Panel A. CRISPR/Cas9 RNPs with sgRNAs targeting both TRAC and TRBC loci were coelectroporated with an ssDNA HDR template encoding murine T-cell receptor α- and β-chains. The HDR template was designed to include 350-nt homology arms targeting exon 1 of the TRAC locus in addition to the following elements: P2A and T2A, self-cleaving peptide inserts; TRBC mouse, mouse TCR-β constant region; TRAC mouse, mouse TCR-α constant region; pGHpA, poly-A tail. The total length of the ssDNA was 2,800 nt. Panel B. Flow cytometric analysis of resulting T-cell populations 10 days after editing using antibodies against mouse TCR-β and human TCR-α/β. Cells in the negative control population (nonedited cells) were positive for the expression of human TCRs. For cells electroporated with RNPs targeting both TRAC and TRBC loci, knockout of the endogenous TCR could be detected. In cells coelectroporated with RNPs combined with the ssDNA HDR template, expression of the murine T-cell receptor α- and β-chains could be detected.
Conclusions
As demonstrated above, the use of ssDNA donor templates for CRISPR-based knockin applications enables more reliable editing with less nonspecific integration and lower cytotoxicity compared to dsDNA templates. However, widespread use of ssDNA has been limited for applications requiring donor templates >200 nt due to the cost and difficulty associated with producing longer ssDNA molecules. The Guide-it Long ssDNA Production System v2 addresses this limitation by enabling the on-demand production of ssDNA ranging from 500–5,000 nt in an error-free and cost-effective manner.
As shown, ssDNA produced with the Guide-it kit was associated with significantly less nonspecific integration and toxicity in comparison to dsDNA when introduced along with Cas9 RNPs into target cells and enabled efficient knockin of multi-kb constructs.
The different cell types were electroporated using a Neon electroporation system according to the following guidelines:
hiPSCs: 1.5 x 105 cells were electroporated in Buffer R with 1 µg of ssDNA or dsDNA in either the presence or absence of 2.25 µg of Cas9 nuclease combined with 0.45 µg of sgRNA. The electroporation conditions were as follows: pulse voltage / pulse width / pulse number = 1100 V / 20 ms / 2 pulses
CD3+ T cells: 3 x 105 cells were electroporated in Buffer T with 0.75 µg of ssDNA in the presence of 1.68 µg of Cas9 nuclease combined with 0.343 µg of sgRNA. The electroporation conditions were as follows: pulse voltage / pulse width / pulse number = 1600 V / 10 ms / 3 pulses
Cytotoxicity of hiPSCs was assessed 48 hr after electroporation by microscopy.
Cytotoxicity of T cells was assessed 24 hr after electroporation by flow cytometry using eBioscience Fixable Viability Dye eFluor 660 (Thermo Scientific Cat# 65-0864-14). Successful KI was detected using the following antibodies: PE anti-human TCR-α/β (Biolegend Cat# 306707) and FITC anti-murine TCR β-chain–APC (Biolegend Cat# 109205).
For the experiments involving HEK293 cells, we used the Guide-it CRISPR/Cas9 System, consisting of a plasmid expressing Cas9 and the sgRNA. The Cas9-sgRNA plasmid was transfected along with the donor templates using Xfect Transfection Reagent. Again, long ssDNA was produced according to the user manual for the Guide-it Long ssDNA Production System v2, and 1 µg of ssDNA or dsDNA was cotransfected along with the Cas9-sgRNA expression plasmid. The transfected cells were subjected to flow cytometry analysis three days posttransfection.
References
Chenet al.,High-frequency genome editing using ssDNA oligonucleotides with zinc-finger nucleases.Nat. Meth.8,753–755 (2011).
Eyquem et al., Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature543(7643), 113–117 (2017).
Roth et al., Reprogramming human T cell function and specificity with nonviral genome targeting. Nature559(7714), 405–409 (2018).
Schober et al., Orthotopic replacement of T-cell receptor a- and b- chains with preservation of near-physiological T-cell function. Nature Biomedical Engineering543(12), 974–984 (2019).