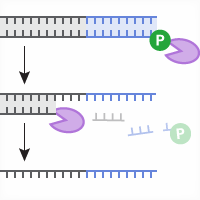
A common application of CRISPR/Cas technology involves engineering gene knockins in which DNA sequences are substituted or inserted at specific genomic loci. In contrast with CRISPR-mediated indels, which result from the error-prone non-homologous end joining (NHEJ) pathway, gene knockins are often engineered via homology-directed repair (HDR), typically through the use of CRISPR reagents (Cas enzyme and guide RNA) in tandem with a DNA template that shares homology with the target site and encodes for the desired modification (Hsu et al., 2014; Figure 1, below). The template used for HDR may be double-stranded DNA (dsDNA, linear or plasmid) or single-stranded DNA (ssDNA), and recent findings have suggested that the repair mechanism varies depending on the type of template used; dsDNA triggers a RAD51-dependent mechanism that mirrors meiotic homologous recombination (HR), whereas HDR involving ssDNA (referred to as single-stranded template repair or SSTR) is RAD51-independent and requires multiple components of the Fanconi Anemia (FA) repair pathway (Richardson et al., 2018).
Figure 1. Engineering CRISPR-mediated gene knockins via homology-directed repair. Following formation of a double-stranded break at a genomic target site via Cas9-gRNA-mediated cleavage, a DNA template sharing homology with the targeted region can trigger homology-directed repair (HDR), resulting in knockin of exogenous sequences encoded in the DNA template (purple).
This approach has been used to engineer small edits as short as single-nucleotide substitutions, and also for the insertion of longer sequences, including fluorescent reporters or gene promoters. Knockin efficiencies can vary widely, depending in part on the species or cell type, the genomic region being targeted, and the size of the insertion. A common method for delivery of the reagents involves coelectroporation of Cas9-gRNA ribonucleoprotein complexes (RNPs) in tandem with DNA HDR templates, because it can be applied to a wide array of cell types and the transience of Cas9-gRNA expression reduces the likelihood of off-target effects. HDR templates consisting of either dsDNA or ssDNA can be used, although ssDNA has been associated with lower toxicity and a reduced likelihood of off-target integration relative to dsDNA (Roth et al., 2018). Smaller ssDNA templates (i.e., ≤200 nt) are typically obtained via oligo synthesis. For longer ssDNA, however, synthesis can be cost prohibitive, and an increasingly popular method is the Guide-it Long ssDNA Production System, which allows for on-demand production of high-quality ssDNA up to 5 kb in length from a double-stranded PCR product via selective digestion of either the sense or antisense strand.
A powerful application of this CRISPR-mediated knockin technology involves site-specific insertion of fluorescent protein sequences to obtain fusion proteins that are expressed under the same promoter and subject to the same regulatory and cell-signaling mechanisms as their endogenous counterparts, as opposed to expressing the fusion protein from a plasmid under a different promoter (e.g., CMV) or from a non-native genomic context via random integration. To demonstrate the suitability of the Guide-it Long ssDNA Production System for this application, we used it to generate two independent hiPS cell lines expressing AcGFP1 fused to the N-termini of SEC61B and Tubulin, respectively.
Results
Design and production of ssDNA templates
Sense and antisense single-stranded DNA (ssDNA) templates for AcGFP1 knockin at the SEC61B and TUBA1A loci were designed to include homology arms of ~350 nt flanking the AcGFP1 sequence (Figure 2), and were produced using the Guide-it Long ssDNA Production System. For comparison purposes, unprocessed dsDNA PCR products corresponding to each HDR template were retained for electroporation in parallel. It has been demonstrated that there is an exponential relationship between homology arm length and knockin efficiency, with ssDNA templates including homology arms 350–700 nt in length providing optimal performance (Li et al., 2018). In our own studies involving hiPSCs, optimal performance was observed with 350-nt homology arms, and the use of longer arms did not substantially increase knockin efficiencies.
Figure 2. Schematics depicting Cas9-sgRNA cleavage sites and ssDNA constructs designed for knockin of AcGFP1 at the SEC61B and TUBA1A loci. The ssDNA constructs included the AcGFP1 coding sequence (~700 nt in length) flanked on either side by ~350-nt homology arms. Binding sites for PCR primers used to detect the inserted AcGFP1 sequence at the SEC61B locus are depicted by blue arrows. As indicated, neither of the ssDNA constructs included a promoter sequence, such that expression of the resulting AcGFP1-tagged fusion proteins would be driven by the respective endogenous promoters.
Electroporation of hiPSCs with ssDNA results in markedly lower toxicity vs. dsDNA
Coelectroporation of Cas9 complexed with single guide RNA (sgRNA) in tandem with HDR templates was performed according to the protocol specified for the Neon system in the Guide-it Recombinant Cas9 (Electroporation-Ready) User Manual. Cas9-sgRNA RNPs were prepared by co-incubating recombinant Cas9 protein with in vitro-transcribed sgRNAs designed to target the SEC61B or TUBA1A locus, and the RNPs were then combined with corresponding HDR templates consisting of either dsDNA or ssDNA (sense or antisense). The various RNP-HDR template combinations and negative-control RNPs without associated HDR templates were then electroporated into ChiPSC18 cells using standard conditions. After 48 hours of culturing, imaging was performed on each cell population to assess the relative toxicity associated with each RNP-HDR template combination (Figure 3). While coelectroporation of ssDNA or dsDNA HDR templates and RNPs yielded noticeable toxicity relative to the application of RNPs alone, the level of toxicity associated with dsDNA was much greater than what was observed for ssDNA. This finding is consistent with the results of previous studies performed by Takara Bio and by others (Roth et al., 2018).
Figure 3. Toxicity comparison for electroporation of dsDNA or ssDNA into iPS cells. ChiPSC18 cells were electroporated with Cas9-sgRNA RNPs targeting the TUBA1A locus in the absence or presence of homology-directed repair (HDR) templates consisting of dsDNA or ssDNA, and then cultured for 48 hours. While electroporation of either DNA template resulted in increased toxicity compared to electroporation of Cas9-sgRNA RNPs alone (as evidenced by lower cell densities), application of ssDNA resulted in greatly reduced toxicity relative to dsDNA.
Analysis of AcGFP1 knockin efficiencies by flow cytometry
Following culturing, the edited populations were analyzed using flow cytometry (Figure 4). In these analyses, the percentage of fluorescent-positive cells is equivalent to the percentage of successful knockins (i.e., knockin efficiency). For each locus, it was observed that the use of sense or antisense ssDNA templates yielded different knockin efficiencies; in the case of SEC61B, the sense template resulted in a higher knockin efficiency than the antisense template (12% vs. 7%, respectively; Figure 4 and data not shown), whereas for TUBA1A, the antisense template provided a higher efficiency (2.7% vs. 1.3%; Figure 4 and data not shown). Currently, there is no clear rule regarding the preferential use of sense or antisense ssDNA for HDR templates longer than 200 nt, but in the case of shorter single-stranded oligodeoxynucleotide (ssODN) templates, template polarity has been found to have an effect on efficiency (Bollen et al., 2018).
Figure 4. Analysis by flow cytometry of AcGFP1 knockin efficiencies at SEC61B and TUBA1A in iPS cells using ssDNA donors. Following electroporation and culturing, edited cell populations and unelectroporated negative control cells were analyzed for AcGFP1 expression by flow cytometry. Measuring the proportion of fluorescent cells in each population allowed for determination of knockin efficiencies: 13% knockin efficiency at SEC61B using a sense ssDNA, and 2.75% efficiency at TUBA1A using an antisense ssDNA.
Detection of monoallelic or biallelic knockin by PCR
One of the most important steps in characterizing edited clonal cell lines involves determining their zygosity. When the inserted sequence is of sufficient length (e.g., AcGFP1), zygosity can be easily assessed via PCR amplification of the target locus using primers that anneal outside of the genomic region that shares homology with the HDR template (Figure 5). In such analyses, a wild-type allele will generate a shorter amplicon than an allele carrying a successful knockin. To determine the zygosity of the different clonal cell lines encoding for an AcGFP1 fusion to SEC61B, genomic DNA was extracted from several of the cell lines and amplified by PCR. Agarose gel analysis of the resulting PCR products indicated that the majority of the clonal cell lines carried biallelic knockins and that comparable editing outcomes were obtained using sense (ss) or antisense (as) ssDNA templates (Figure 5).
Figure 5. Detection of monoallelic or biallelic knockin by PCR amplification of the target site. Genomic DNA was extracted and analyzed by PCR using forward and reverse primers designed to anneal outside the genomic region sharing homology with the HDR template. The AcGFP1 and homology arm sequences encoded in the ssDNA templates are shown in green and light blue, respectively. The PCR primers were designed such that presence or absence of an inserted AcGFP1 sequence would yield PCR products of different sizes (1.9 kb or 1.2 kb, respectively). Gel electrophoresis of PCR products confirmed the occurrence of monoallelic AcGFP1 knockins, represented by double bands (e.g., ssDNA-as, Lane C5), and biallelic AcGFP1 knockins, represented by single bands corresponding to the larger product size (e.g., ssDNA-as, Lane C1). Two wild-type clones lacking any copies of the AcGFP1 insert were also identified (ssDNA-as, Lane C10 and ssDNA-ss, Lane C11).
Analysis of AcGFP1-tagged protein localization by fluorescence microscopy
Following isolation and expansion, AcGFP1-positive clones were imaged using fluorescence microscopy (Figure 6). In each clonal population, AcGFP1 fluorescence matched the canonical localization pattern of the protein encoded by the intended genomic target: targeting of the SEC61B locus yielded AcGFP1 expression in endosomal compartments, while targeting of the TUBA1A locus resulted in fluorescent labelling of microtubules. These results suggest that electroporation of RNPs and ssDNA templates targeting SEC61B or TUBA1A resulted in successful insertion of AcGFP1 at the corresponding locus, such that the resulting fusion proteins localize in the same manner as their untagged, endogenous counterparts.
Figure 6. Analysis of AcGFP1 localization in clonal cell lines following editing. AcGFP1-positive clonal cell lines derived from each edited population were fixed and counterstained with DAPI and analyzed by fluorescence microscopy. In each case, AcGFP1 localized in a manner consistent with the function of the endogenous protein: endosomal compartments (SEC61B locus) and microtubules (TUBA1A locus), respectively.
Conclusions
The combination of CRISPR/Cas technology with the application of DNA templates for homology-directed repair has given rise to a powerful method for precise genome editing that can be employed in a wide array of experimental contexts. However, the efficiency of this approach varies greatly, depending on the cell type(s) and the genomic loci being targeted, and a persistent challenge involves delivering HDR templates in a manner that minimizes toxicity and the likelihood of off-target integration. Here we have presented data demonstrating that electroporation of Cas9-sgRNA RNPs in tandem with ssDNA generated using the Guide-it Long ssDNA Production System enables efficient and accurate knockin of long sequences in hiPSCs with minimal toxicity. Given the relative simplicity and cost-effectiveness of the approach presented here, we anticipate that it will continue to drive greater adoption and application of CRISPR/Cas9 technology for precise genome editing.
References
Bollen et al., How to create state-of-the-art genetic model systems: strategies for optimal CRISPR-mediated genome editing. Nuc. Acids Res.46, 6435–6454 (2018).
Hsu et al., Development and applications of CRISPR-Cas9 for genome engineering. Cell157, 1262–1278 (2014).
Li et al., Design and specificity of long ssDNA donors for CRISPR-based knockin. bioRxiv, https://doi.org/10.1101/178905 (2017).
Richardson et al., CRISPR-Cas9 genome editing in human cells occurs via the Fanconi anemia pathway. Nat. Gen.50, 1132–1139 (2018).
Roth et al., Reprogramming human T cell function and specificity with non-viral genome targeting. Nat. Lett.559, 405–409 (2018).