

Liver metabolic function, dissecting one cell at a time Q&A
Single-cell RNA-seq can help elucidate the role of pathogenic cell populations in the development and progression of chronic diseases.
In this webinar Dr. Martinez-Jimenez and Dr. Deligiannis present their findings on the practical application of snRNA-seq2, a robust and reliable method for the analysis of the nuclear transcriptome from frozen tissues developed by customizing the Takara Bio SMART-Seq Single Cell Kit . The cost-efficient miniaturization of snRNA-seq2 using liquid handling robots (i.e., the mosquito HV genomics system, SPT Labtech) is also discussed.
Continue reading for insights from the Q&A session or register to watch the on-demand webinar.
How can we gain access to the scripts for snRNA-seq2?
The Jupyter Notebook code for analysis is available for download here and in Zenodo. The data is now publicly available here and here.
How can we gain access to the scripts for the SMART-Seq Single Cell Kit on mosquito HV?
To receive the mosquito scripts for cDNA synthesis and bead clean-ups, please get in touch with the SPT Labtech Field Applications Team.
If working with multiple cells per well, which kit would you recommend?
In this case, we would recommend the the SMART-Seq v4 Kit.
The SMART-Seq v4 Kit (SSv4) and the SMART-Seq v4 PLUS Kit (SSv4 PLUS) are the fourth generation of our SMARTer ultra-low solutions and our most sensitive kits for ultra-low inputs. These kits use oligo(dT) priming to generate high-quality, full-length cDNA directly from multiple intact cells (up to 1,000 cells) or 10 pg–10 ng of total RNA.
Can you distinguish between 4n bi- and 4n mono-nucleated cells with the snRNA-seq methodology?
With the transcriptional profile alone, it is difficult to distinguish between 2n nuclei from mononucleated hepatocytes and 2n nuclei from tetraploid binucleated hepatocytes (2n x 2).
Technologies such as RNAScope (in situ hybridization assay for detection of target RNA within intact cells) can be used to distinguish this hepatocyte ploidy. Spatial transcriptomics approaches in the future will be very useful to address these questions genome-wide.
Have you explored differences in splicing variants between polyploid hepatocytes?
Taking advantage of the full-length transcript information that snRNA-seq2 offers, we are now investigating the potential splicing variant differences between hepatocyte nuclei.
Has this strategy been used with other tissues?
In our lab we have used snRNA-seq2 method with liver, gut, brain and muscle tissue.
What was the ratio of volume of lysis buffer/volume of tissue sample used in the study?
For the single nuclei isolation, the protocol described by Krishnaswami et al. was followed with several modifications.
In summary, fresh frozen liver tissues (~3 mm3) were homogenized and the final nuclei suspension was used for FACS sorting of 1 nuclei per well containing 0.9 µL final volume of the Takara Bio lysis buffer.
Do you have optimized protocols for extracting nuclei from human tissues (such as lung)?
We have used the nuclei isolation protocol described in snRNA-seq2 with various tissues from mice, including lung and we obtain a clean suspension. Similarly, we have used it for human tissues, liver and bone marrow but not with human lung. We are positive that it would work although it might require additional optimization to increase the purity of the single-nuclei suspension.
Were differences in ploidy status relevant in stellate cells in fibrotic conditions?
We have not explored the differences on stellate cell ploidy status. We focused on hepatocytes in this study.
How does ploidy correlate with oxidative or glycolytic metabolism?
According to analysis via Gene Ontology between 2n and 4n hepatocytes, an enrichment of genes related to lipid and xenobiotic metabolism as well as oxidative stress pathways is oberserved in 4n hepatocytes. Other pathways can be explored and investigated under different treatments and conditions.
When performing snRNA-seq2 method, could you potentially identify nuclei from binuclear polyploid cells using bioinformatical tools?
Starting from single-nuclei suspensions, we can only distinguish DNA content per nuclei using Hoescht and FACs sorting. We found no significant differences in the mRNA levels between 2n nuclei in diploid and tetraploid binucleated cells using RNAscope, and those populations are not able to be distinguished by snRNA-seq2 computationally.
Does the Takara Bio SMART-Seq Single Cell Kit allow deep characterization of nuclei isolated from fresh tissues?
Both fresh and frozen tissues can be used performing the with the snRNA-seq2 using the SMART-Seq Single Cell Kit. When possible, fresh tissue and whole cell approaches might be preferred, depending on the experimental design, dissociation methods, and accessibility to fresh samples.
Is polyploidy related to liver cell regeneration?
There are several recent publications using lineage tracing strategies to investigate the regenerative potential of tetraploid hepatocytes (Chen F, et al., 2020; Sun T, et al. 2020; Matsumoto T, et al., 2020).
Their work and our work conclude that, both 2n and 4n can contribute to liver regeneration, although 4n cells need to divide into 2n first.
Does the snRNA-seq2 method work for low-input/low-quality samples (e.g. single cells collected from laser microdissection)?
Although we have not used this methodology for single cell/nuclei RNAseq from laser microdissection, the addition of lysis buffer 2 is compatible with downstream SMARTer chemistry. As long as a strategy for placing the cell in wells with lysis buffer is in place, the cDNA generation using the presented pipeline should be straight-forward.
Do you use FACS just for sorting 2n and 4n cells, or it is also used for purification purposes?
For our snRNA-seq2 approach, we use FACS sorting to address ploidy levels in our nuclei suspension and sort single nuclei into wells. The FACS sorting step though is not always necessary for downstream RNAseq, as long as the suspension is clean enough. For single-cell RNAseq based on well-plates, FACS sorting or other instruments for dispensing single-cells/nuclei into weeks will be needed.
Do you miss relevant extranuclear transcripts with snRNA-seq2?
Using snRNA-seq2, only the nuclear mRNA is captured, loosing partial information about the mature cytoplasmic mRNAs. The high correlation between nuclear and whole-cell transcriptome shows that the analysis of nuclear transcripts is a robust and reliable approach for cell-type identification.
Do male and female mice show the same percentage of polyploidies in their livers?
We have not investigated gender bias in hepatocyte ploidy. It will certainly be an interesting question to explore.
Is the data on immunohistochemistry available online?
Our BioRxiv manuscript was recently accepted in Nature Communications, and all data will be publicly available shortly.
Are nuclei from a binuclear cell transcriptionally similar?
In young C57Bl6/J, the expression levels of mononucleated and binucleated hepatocytes are conditioned by their position in the hepatic lobule (confirmed with RNAScope). The global expression profiles of 2n nuclei and 4n nuclei is very similar and they are not easily distinguished without previous knowledge on their genome content (e.g. by DAPI or Hoescht).
Could you detect interference of cryopreservation agents like RNAlater or others with your protocol?
We have not tried our pipeline with RNAlater or other cryopreservation methods. We do not know if cryopreservation reagents would have a major impact in the in nuclear transriptome.
How do you avoid DNA contamination?
In our data, we are not able to detect high DNA contamination sequences. In the case of high DNA content, we can follow the manufacturer's suggestion to pre-treat the samples with DNAse I.
What is the best way to improve nuclei quality and integrity after sort?
To acquire clean nuclei suspension, we use an OptiPrep gradient and inspect the quality under a microscope. For further improvements, washing and filtering steps can be used. To maintain integrity, the appropriate nuclei storage buffer needs to be used for each tissue and all manipulations needs to be done with caution regarding shearing forces.
Is there a difference in RNA expression between 2n and 4n in NASH and HCC?
We are now further investigating the changes in the transcriptome of human chronic liver diseases and whether the percentages of 2n to 4n change as well.
SMART-Seq Single Cell Kit resources
SSsc for scRNA-seq
Generate high-quality, full-length RNA-seq libraries from single cells (e.g., PBMCs) and nuclei.
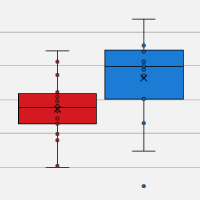
Highest sensitivity for single-cell mRNA-seq
View data on the SMART-Seq Single Cell Kit's superior sensitivity and reproducibility for single-cell and nuclei applications.
Takara Bio USA, Inc.
United States/Canada: +1.800.662.2566 • Asia Pacific: +1.650.919.7300 • Europe: +33.(0)1.3904.6880 • Japan: +81.(0)77.565.6999
FOR RESEARCH USE ONLY. NOT FOR USE IN DIAGNOSTIC PROCEDURES. © 2025 Takara Bio Inc. All Rights Reserved. All trademarks are the property of Takara Bio Inc. or its affiliate(s) in the U.S. and/or other countries or their respective owners. Certain trademarks may not be registered in all jurisdictions. Additional product, intellectual property, and restricted use information is available at takarabio.com.
